Sonication Chromatin Immunoprecipitation Protocol
Specific for product: SimpleChIP® Plus Sonication Chromatin IP Kit #56383.
I. Tissue Cross-linking and Sample Preparation
When harvesting tissue, remove unwanted material from the sample, such as fat and necrotic material. Tissue can be processed and cross-linked immediately or frozen on dry ice for processing later. For optimal ChIP results, use 25 mg of tissue for each immunoprecipitation to be performed. An additional 5 mg of tissue should be processed for Analysis of Chromatin Digestion and Concentration and for use as input chromatin (Section IV). The chromatin yield does vary between tissue types, and some tissues may require more than 25 mg for each immunoprecipitation.
One chromatin preparation is defined as 100 to 150 mg of tissue. This recommended amount of tissue accounts for potential low yield with some tissue types and also ensures efficient chromatin fragmentation during sonication. Please see Appendix A for more information regarding the expected chromatin yield for different types of tissue.
Before starting:
- Remove and warm 200X Protease Inhibitor Cocktail (PIC) #7012 and Glycine Solution (10X) #7005. Make sure PIC is completely thawed.
- Prepare 3 ml of Phosphate Buffered Saline (PBS) + 15 µl 200X PIC per chromatin preparation and place on ice.
- Prepare 1 ml of 1X ChIP Sonication Cell Lysis Buffer (0.5 ml 2X ChIP Sonication Cell Lysis Buffer #96529 + 0.5 ml water) + 5 µl 200X PIC per chromatin preparation and place on ice.
- Prepare 28 µl of 37% formaldehyde per chromatin preparation and keep at room temperature. Alternatively, 62.5 µl of 16% methanol-free formaldehyde can be used. Use fresh formaldehyde that is not past the manufacturer's expiration date.
- Weigh the fresh or frozen tissue sample. Use 100 to 150 mg of tissue per chromatin preparation.
- Place tissue sample in a petri dish and mince into 1-2 mm cubes using a clean scalpel or razor blade. Keep dish on ice. It is important to keep the tissue cold to avoid protein degradation.
- Transfer minced tissue to a 15 ml conical tube and add 1 ml of ice-cold PBS + PIC per chromatin preparation.
- To crosslink proteins to DNA, add 28 µl of 37% formaldehyde or 62.5 µl of 16% methanol-free formaldehyde per 1 ml of PBS + PIC and keep at room temperature for at least 10 min. Final formaldehyde concentration is 1%. For histone modification ChIP, 10 min fixation is sufficient; however, for transcription factor ChIP, we recommend fixation for 10 to 30 min, and for transcription cofactor ChIP, we recommend fixation for 30 min (see Figure 7 in Appendix B).
- Stop cross-linking by adding 100 µl of 10X Glycine #7005 per 1 ml of PBS + PIC. Mix and incubate on ice for 5 min.
- Centrifuge tissue at 1,200 x g for 5 min at 4°C.
- Remove supernatant and wash with 1 ml of ice-cold PBS + PIC per chromatin preparation.
- Centrifuge at 1,200 x g for 5 min at 4°C.
- Repeat step 7 and 8 one additional time.
- Remove supernatant and resuspend tissue in 1 ml 1X ChIP Sonication Cell Lysis Buffer + PIC per chromatin preparation.
- Transfer tissue suspension to a Dounce homogenizer using a cut pipet tip.
- Use tight fitting pestle (Type A) to disaggregate tissue pieces with 20 strokes or until no chunks of tissue are observed.
- Transfer cell suspension to a 1.5 ml tube and immediately proceed to Nuclei Preparation and Chromatin Fragmentation (Section III).
II. Cell Culture Cross-linking and Sample Preparation
For optimal ChIP results, use approximately 4 x 106 cells for each immunoprecipitation to be performed. For HCT 116 cells, this is equivalent to 1/3 of a 15 cm culture dish containing cells that are 90% confluent in 20 ml of growth medium. An additional 1 x 106 cells should be processed for Analysis of Chromatin Digestion and Concentration and for use as input chromatin (Section IV).
One chromatin preparation is defined as 1 x 107 to 2 x 107 cells. This recommended cell number accounts for potential low yield with some cell types and also ensures efficient chromatin fragmentation during sonication.
Before starting:
- Remove and warm 200X Protease Inhibitor Cocktail (PIC) #7012 and Glycine Solution(10X) #7005. Make sure PIC is completely thawed.
- Prepare 2 ml of Phosphate Buffered Saline (PBS) + 10 µl 200X PIC per 15 cm dish to be processed and place on ice.
- Prepare 40 ml of PBS per 15 cm dish to be processed and place on ice.
- Prepare 540 µl of 37% formaldehyde per 15 cm dish of cells to be processed and keep at room temperature. Alternatively, 1.25 ml of 16% methanol-free formaldehyde can be used. Use fresh formaldehyde that is not past the manufacturer's expiration date.
- Prepare 1 ml of 1X ChIP Sonication Cell Lysis Buffer (0.5 ml 2X ChIP Sonication Cell Lysis Buffer #96529 + 0.5 ml water) + 5 µl 200X PIC per chromatin preparation.
- To crosslink proteins to DNA, add 540 µl of 37% formaldehyde or 1.25 ml of 16% methanol-free formaldehyde to each 15 cm culture dish containing 20 ml medium. Swirl briefly to mix and incubate 10 min at room temperature. Final formaldehyde concentration is 1%. Addition of formaldehyde may result in a color change of the medium.
- Add 2 ml of 10X glycine to each 15 cm dish containing 20 ml medium, swirl briefly to mix, and incubate 5 min at room temperature. Addition of glycine may result in a color change of the medium.
- For suspension cells:
- Transfer cells to a 50 ml conical tube, centrifuge 1,000 x g for 5 min at 4°C, and wash pellet two times with 20 ml ice-cold PBS. Remove PBS and proceed to step 3b. Alternatively, cell pellets can be frozen on dry ice and stored at -80°C for later use.
- Resuspend up to 2 x 107 cells per 1 ml of 1X ChIP Sonication Cell Lysis Buffer + PIC per chromatin preparation, and immediately proceed to Nuclei Preparation and Chromatin Fragmentation (Section III).
- For adherent cells:
- Remove media and wash cells two times with 20 ml ice-cold 1X PBS, completely removing wash from culture dish each time.
- Add 2 ml ice-cold PBS + PIC to each 15 cm dish. Scrape cells into cold buffer. Combine cells from all culture dishes into one 15 ml conical tube.
- Centrifuge cells at 1,000 x g for 5 min at 4°C. Remove PBS and proceed to step 4d. Alternatively, cell pellets can be frozen on dry ice and stored at -80°C for later use
- Resuspend up to 2 x 107 cells per 1 ml of 1X ChIP Sonication Cell Lysis Buffer + PIC per chromatin preparation and immediately proceed to Nuclei Preparation and Chromatin Fragmentation (Section III).
III. Nuclei Preparation and Chromatin Fragmentation
One chromatin preparation is defined as 100 to 150 mg of tissue or 1 x 107to 2 x 107tissue culture cells. Multiple chromatin preparations can be performed simultaneously, as long as the amounts of buffers are scaled appropriately and sonication is performed on 1 ml samples. The number of cells and volume of sample used for sonication is critical for generation of appropriately sized chromatin fragments.
Before starting:
- Remove and warm 200X Protease Inhibitor Cocktail (PIC) #7012. Make sure it is completely thawed prior to use.
- Prepare 1 ml of 1X ChIP Sonication Cell Lysis Buffer (0.5 ml 2X ChIP Sonication Cell Lysis Buffer #96529 + 0.5 ml water) + 5 µl 200X PIC per chromatin preparation.
- Prepare 1 ml ChIP Sonication Nuclear Lysis Buffer #28778 + 5 µl 200X PIC per chromatin preparation.
- Incubate cell suspension in 1X ChIP Sonication Cell Lysis Buffer + PIC from Section I or Section II on ice for 10 min.
- Pellet cells at 5,000 x g for 5 min at 4°C. Remove supernatant and resuspend pellet a second time in 1 ml ice-cold 1X ChIP Sonication Cell Lysis Buffer + PIC per chromatin preparation.
- Incubate cell suspension on ice for 5 min. Pellet cells at 5,000 x g for 5 min at 4°C. Please note that cross-linked cells may not be completely lysed until sonication.
- Resuspend cells in 1 ml ice-cold ChIP Sonication Nuclear Lysis Buffer #28778 + PIC per chromatin preparation and incubate on ice for 10 min. Transfer 1 ml of cell suspension to an appropriately sized tube for sonication. Please note that cross-linked cells and nuclei may not be completely lysed until sonication.
- Fragment chromatin by sonication. Sonication conditions may need to be determined empirically by testing different sonicator power settings and durations of sonication. Optimal sonication conditions will generate chromatin in which 60 to 90% of the chromatin fragments are smaller than 1kb. Longer cross-linking times may decrease the proportion of fragments being smaller than 1 kb to 30 to 60% (see Figure 7 in Appendix B and Figure 8 in Appendix C). Use the minimal number of sonication cycles required to generate the desired length of chromatin fragments, as over-sonication can result in reduction or loss of signal due to harsh treatment of the chromatin.
- For each sonication sample, we recommend using 100-150 mg of tissue or 1 x 107to 2 x 107cells per 1 ml ChIP Sonication Nuclear Lysis Buffer. Sonicating in larger volumes and/or using greater concentration of cells will decrease the efficiency of chromatin fragmentation.
- Using a Branson Digital Sonifier D250 probe sonicator with a 1/8-inch Micro Tip, 8 min of 1 sec on/1 sec off sonication cycle (4 min of sonication time) at 50% amplitude typically gives good fragmentation and chromatin IP efficiency.
- Be sure to keep the chromatin sample cool during sonication by keeping the tube containing the chromatin in an ice water bath during and in between the sonication steps. Do not allow the probe to touch the bottom or wall of the tube. Stop sonication and adjust the position of tube if chromatin sample foams during sonication.
- Clarify lysates by centrifugation at 21,000 x g in a microcentrifuge for 10 min at 4°C.
- Transfer supernatant to a new tube. This is the cross-linked chromatin preparation, which can be used immediately for immunoprecipitation, or stored at -80°C until further use. Remove 50 µl of the chromatin preparation for Analysis of Chromatin Digestion and Concentration (Section IV).
IV. Analysis of Chromatin Fragmentation and Concentration (Recommended Step)
- To the 50 µl chromatin sample (from Step 7 in Section III), add 100 µl nuclease-free water, 6 µl 5 M NaCl #7010, and 2 µl RNAse A#7013. Vortex to mix and incubate samples at 37°C for 30 min.
- To each RNase A-digested sample, add 2 µl Proteinase K #10012. Vortex to mix and incubate samples at 65°C for 2 hr.
- Purify DNA from samples using DNA purification spin columns as described in Section VII.
- After purification of DNA, remove a 10 µl sample and determine DNA fragment size by electrophoresis on a 1% agarose gel. A DNA smear ranging from 200 bp to several kb is expected (see Figure 7 in Appendix C). Approximately 60 to 90% of total DNA fragments should be less than 1 kb. Refer to Appendix B for optimization of sonication conditions.
- Determine DNA concentration using a spectrophotometer. Ideally, DNA concentration should be between 50 and 200 µg/ml.
V. Chromatin Immunoprecipitation
For optimal ChIP results, use approximately 5 to 10 µg of sonicated, cross-linked chromatin (as determined in Section IV) per immunoprecipitation. This should be roughly equivalent to a single 100 µl IP prep from 25 mg of disaggregated tissue or 4x106 tissue culture cells. Typically, 100 µl of digested chromatin is diluted into 400 µl 1X ChIP Buffer prior to the addition of antibodies. However, if more than 100 µl of chromatin is required per IP, the cross-linked chromatin preparation must be diluted into 1X ChIP buffer at a dilution ratio of 1:4. No additional protein G magnetic beads are necessary in this case, although prolonged incubation time with beads is helpful.
Before starting:
- Remove and warm 200X Protease Inhibitor Cocktail (PIC) #7012. Make sure PIC is completely thawed.
- Remove and warm 10X ChIP Buffer #7008 and ensure SDS is completely in solution.
- Thaw fragmented chromatin preparation (from Step 7 in Section III) and place on ice.
- Prepare low salt wash: 3 ml 1X ChIP Buffer (300 µl 10X ChIP Buffer #7008 + 2.7 ml water) per immunoprecipitation. Keep on ice.
- Prepare high salt wash: 1 ml 1X ChIP Buffer (100 µl 10X ChIP Buffer #7008 + 900 µl water) + 70 µl 5M NaCl #7010 per immunoprecipitation. Keep on ice.
- In one tube, prepare enough 1X ChIP Buffer + PIC for the dilution of sonicated chromatin into the desired number of immunoprecipitations. For each immunoprecipitation, dilute chromatin (5-10 µg of DNA) with 1X ChIP Buffer + PIC into a total of 500 µl. For efficient immunoprecipitation, chromatin must be diluted into 1 x ChIP buffer at a dilution factor of 1:4 or greater. Place mix on ice. When determining the number of immunoprecipitations, remember to include the positive control Histone H3 (D2B12) XP® Rabbit mAb#4620 and negative control Normal Rabbit IgG antibody #2729 samples.
- Remove a 10 µl sample of the diluted chromatin and transfer to a microfuge tube. This is your 2% Input Sample, which can be stored at -20°C until further use (Step 1 in Section VI).
- For each immunoprecipitation, aliquot 500 µl the diluted chromatin into a 1.5 ml microcentrifuge tube and add the immunoprecipitating antibody. The amount of antibody required per IP varies and should be used according to the manufacturers' recommendations. For the positive control Histone H3 (D2B12) XP® Rabbit mAb#4620, add 10 µl to the IP sample. For the negative control Normal Rabbit IgG #2729, add 1 µl (1 µg) to 2 µl (2 µg) to the IP sample. Incubate IP samples 4 hr to overnight at 4°C with rotation.
NOTE: Most antibodies from Cell Signaling Technology work optimally between 1 and 2 µg per IP sample. In the case where there are multiple samples with varying concentrations, it is best to match the negative control Normal Rabbit IgG #2729 to the highest antibody concentration.
- Resuspend ChIP-Grade Protein G Magnetic Beads #9006 by gently vortexing. Immediately add 30 µl of Protein G Magnetic Beads to each IP reaction and incubate for 2 hr at 4°C with rotation. Alternatively, beads can be added to the overnight antibody incubation in Step 3, but this may increase background signal.
- Pellet Protein G Magnetic Beads in each immunoprecipitation by placing the tubes in a magnetic separation rack. Wait 1 to 2 min for solution to clear and then carefully remove supernatant.
- Wash Protein G Magnetic Beads by adding 1 ml of low salt wash to the beads and incubate at 4°C for 5 min with rotation. Repeat steps 6 and 7 two additional times for a total of 3 low salt washes.
- Add 1 ml of high salt wash to the beads and incubate at 4°C for 5 min with rotation.
- Pellet Protein G Magnetic Beads in each immunoprecipitation by placing the tubes in a magnetic separation rack. Wait 1 to 2 min for solution to clear and then carefully remove supernatant. Immediately proceed to Section VI.
VI. Elution of Chromatin from Antibody/Protein G Magnetic Beads and Reversal of Cross-links
Before starting:
- Remove and warm 2X ChIP Elution Buffer #7009 in a 37°C water bath and ensure SDS is in solution.
- Set a water bath or thermomixer to 65°C.
- Prepare 150 µl 1X ChIP Elution Buffer (75 µl 2X ChIP Elution Buffer #7009 + 75 µl water) for each immunoprecipitation and the 2% input samples.
- Add 150 µl of the 1X ChIP Elution Buffer to the 2% input sample tubes and set aside at room temperature until Step 7.
- Add 150 µl 1X ChIP Elution Buffer to each IP sample.
- Elute chromatin from the antibody/Protein G Magnetic Beads for 30 min at 65°C with gentle vortexing (1,200 rpm). A thermomixer works best for this step; however a 65°C water bath with frequent mixing is also sufficient. Alternatively, elutions can be performed at room temperature with rotation, but may not be as complete.
- Briefly centrifuge samples at 10,000 x g for 10 sec to remove evaporated sample from the microcentrifuge tube cap.
- Pellet Protein G Magnetic Beads by placing the tubes in a magnetic separation rack and wait 1 to 2 min for solution to clear.
- Carefully transfer eluted chromatin supernatant to a new tube.
- To all tubes, including the 2% input samples from Step 1, reverse cross-links by adding 6 µl 5M NaCl #7010 and 2 µl Proteinase K #10012, and incubate 2 hr at 65°C. This incubation can be extended overnight.
- Immediately proceed to Section VII. Alternatively, samples can be stored at -20°C. However, to avoid formation of a precipitate, be sure to warm samples to room temperature before adding DNA Binding Buffer #10007 (Section VII, Step 1).
VII. DNA Purification Using Spin Columns:
Before starting:
- Add 24 ml of ethanol (96-100%) to DNA Wash Buffer #10008 before use. This step only has to be performed once prior to the first set of DNA purifications.
- Prepare one DNA purification spin column and collection tube #10010 for each DNA sample from Section VI.
- Add 750 µl DNA Binding Buffer #10007 to each DNA sample and vortex briefly.
- 5 volumes of DNA Binding Buffer should be used for every 1 volume of sample.
- Transfer 450 µl of each DNA sample from Step 1 to a DNA spin column in collection tube.
- Centrifuge at 14,000 rpm in a microcentrifuge for 30 sec.
- Remove the spin column from the collection tube and discard the liquid. Replace spin column in the collection tube.
- Transfer the remaining 450 µl of each DNA sample from Step 1 to the spin column in collection tube. Repeat Steps 3 and 4.
- Add 750 µl of DNA Wash Buffer #10008 to the spin column in collection tube.
- Centrifuge at 14,000 rpm in a microcentrifuge for 30 sec.
- Remove the spin column from the collection tube and discard the liquid. Replace spin column in the collection tube.
- Centrifuge at 14,000 rpm in a microcentrifuge for 30 sec
- Discard collection tube and liquid. Retain spin column.
- Add 50 µl of DNA Elution Buffer #10009 to each spin column and place into a clean 1.5 ml microcentrifuge tube.
- Centrifuge at 14,000 rpm in a microcentrifuge for 30 sec to elute DNA.
- Remove and discard DNA spin column. Eluate is now purified DNA. Samples can be stored at -20°C.
VIII. Quantification of DNA by PCR:
Recommendations:
- Use filtered pipette tips to minimize risk of contamination.
- The control primers included in the kit are specific for the human or mouse RPL30 gene (#7014 or #7015) and can be used for either standard PCR or quantitative real-time PCR. If the user is performing ChIP from another species, it is recommended that the user design the appropriate specific primers to DNA and determine the optimal PCR conditions.
- A Hot-Start Taq polymerase is recommended to minimize the risk of nonspecific PCR products.
- PCR primer selection is critical. Primers should be designed with close adherence to the following criteria:
| Primer length: |
24 nucleotides |
| Optimum Tm: |
60°C |
| Optimum GC: |
50% |
| Amplicon size: |
150 to 200 bp (for standard PCR) |
|
80 to 160 bp (for real-time quantitative PCR) |
Standard PCR Method:
- Label the appropriate number of PCR tubes or PCR plates compatible with the model of PCR machine to be used. These should include the 2% input sample, the positive control histone H3 sample, the negative control normal rabbit IgG sample, and a tube with no DNA to control for DNA contamination.
- Add 2 µl of the appropriate DNA sample to each tube.
- Prepare a master reaction mix as described below, making sure to add enough reagent for two extra tubes to account for loss of volume. Add 18 µl of master mix to each reaction tube.
| Reagent |
Volume for 1 PCR Reaction (18 µl) |
| Nuclease-free H2O |
12.5 µl |
| 10X PCR Buffer |
2.0 µl |
| 4 mM dNTP Mix |
1.0 µl |
| 5 µM RPL30 Primers |
2.0 µl |
| Taq DNA Polymerase |
0.5 µl |
- Start the following PCR reaction program:
- Initial Denaturation 95°C 5 min
- Denature 95°C 30 sec
- Anneal 62°C 30 sec
- Extension 72°C 30 sec
- Repeat Steps b-d for a total of 34 cycles.
- Final Extension 72°C 5 min
- Remove 10 µl of each PCR product for analysis by 2% agarose gel or 10% polyacrylamide gel electrophoresis with a 100 bp DNA marker. The expected size of the PCR product is 161 bp for human RPL30 #7014 and 159 bp for mouse RPL30 #7015.
Real-Time Quantitative PCR Method:
- Label the appropriate number of PCR tubes or PCR plates compatible with the model of PCR machine to be used. PCR reactions should include the positive control histone H3 sample, the negative control normal rabbit IgG sample, a tube with no DNA to control for contamination, and a serial dilution of the 2% input chromatin DNA (undiluted, 1:5, 1:25, 1:125) to create a standard curve and determine the efficiency of amplification.
- Add 2 µl of the appropriate DNA sample to each tube or well of the PCR plate.
- Prepare a master reaction mix as described below. Setup 2-3 replicates for each PCR reaction. Add enough reagents to account for loss of volume. Add 18 µl of reaction mix to each PCR reaction tube or well. Reagent Volume for 1 PCR Reaction (18 µl)
| Nuclease-free H2O |
6 µl |
| 5 µM RPL30 Primers |
2 µl |
| SimpleChIP® Universal qPCR Master Mix #88989 |
10 µl |
- Start the following PCR reaction program:
| a. |
Initial Denaturation |
95°C 3 min |
| b. |
Denature |
95°C 15 sec |
| c. |
Anneal and Extension: |
60°C 60 sec |
| d. |
Repeat steps b and c for a total of 40 cycles. |
|
- Analyze quantitative PCR results using the software provided with the real-time PCR machine. Alternatively, one can calculate the IP efficiency manually using the Percent Input Method and the equation shown below. With this method, signals obtained from each immunoprecipitation are expressed as a percent of the total input chromatin.
Percent Input = 2% x 2(C[T] 2%Input Sample - C[T] IP Sample)
C[T] = CT = Average threshold cycle of PCR reaction
IX. NG-Sequencing Library Construction
The immuno-enriched DNA samples prepared with this kit are directly compatible with ChIP-seq. For downstream NG-sequencing DNA library construction, use a DNA library preparation protocol or kit compatible with your downstream sequencing platform. For sequencing on Illumina® platforms, we recommend DNA Library Prep Kit for Illumina® (ChIP-seq, CUT&RUN) #56795 and its associated index primers Multiplex Oligos for Illumina® (Single Index Primers) (ChIP-seq, CUT&RUN) #29580 or Multiplex Oligos for Illumina® (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538.
Recommendations:
- For transcription factor or co-factor ChIP-seq, use at least 5 ng of ChIP-enriched DNA and amplification of the adaptor-ligated DNA with 10 cycles of PCR.
- For total histone and histone modifications, or input samples, start with 50 ng of ChIP-enriched DNA and amplification of the adaptor-ligated DNA with 6 cycles of PCR.
- For library construction of ChIP-enriched DNA for all target types, perform cleanup of adaptor-ligated DNA without size selection.
- After DNA library construction, check the DNA library for presence of adaptor dimers (~140 bp) using an Agilent High Sensitivity DNA Kit (Agilent Technologies, Cat# G2938-90322), or by agarose gel electrophoresis with 50-100 ng DNA on a 2% agarose TAE gel. If adaptor dimers are present, repeat cleanup of PCR amplified material.
- The quality of the library can also be confirmed using qPCR and primer sets to known positive and negative target loci. Positive primer pairs should still give the same high signal compared to negative primers as seen in the original qPCR analysis of ChIP-enriched DNA.
- After final cleanup and quality checks, prepare final purified library samples at 2-10 nM for high throughput sequencing.
Appendix A: Expected Chromatin Yield
When harvesting cross-linked chromatin from tissue samples, the yield of chromatin can vary significantly between tissue types. The table to the right provides a range for the expected yield of chromatin from 100 mg of tissue compared to 2 x 107 HCT 116 cells, and the expected DNA concentration, as determined in Section IV of the protocol. For optimal ChIP results, we recommend DNA Library Prep Kit for Illumina® (ChIP-seq, CUT&RUN) #56795 and its associated index primers Multiplex Oligos for Illumina® (Single Index Primers) (ChIP-seq, CUT&RUN) #29580 or Multiplex Oligos for Illumina® (Dual Index Primers) (ChIP-seq, CUT&RUN) #47538.
| Tissue/Cell |
Total Chromatin Yield |
Expected DNA Concentration |
| Liver |
50 µg per 100 mg tissue |
150 µg/ml |
| Brain |
25 µg per 100 mg tissue |
50 µg/ml |
| Heart |
105 µg per 100 mg tissue |
20 µg/ml |
| HCT 116 |
100-150 µg per 2 x 107 cells |
100-150 µg/ml |
Appendix B: Optimization of Formaldehyde Fixation
Transcription factors and cofactors bind to chromatin DNA more loosely than histone proteins. As a result, they tend to dissociate from chromatin during sonication. Additional fixation time can result in increased capture of transcription factors and cofactors in the ChIP assay, especially with tissue samples. As shown in Figure 7, increasing the fixation time from 10 min to 30 min may reduce chromatin fragmentation size (left panel), but it significantly enhances enrichment of both cofactors RING1B and SUZ12 in heart tissue, as indicated by ChIP-qPCR (middle and right panels).
Typically, 10 min fixation is sufficient for histone modification ChIP with both cell and tissue samples, whereas transcription factors and cofactors may require additional fixation up to 30 min, especially with tissue samples.
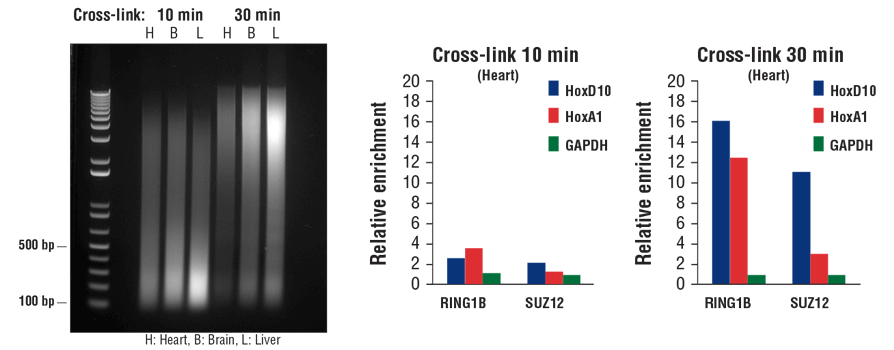
FIGURE 7. Mouse heart (H), brain (B), and liver (L) were cross-linked for 10 min or 30 min, as indicated (left panel). The chromatin was prepared and sonicated, DNA was purified and 20 µl was separated by electrophoresis on a 1% agarose gel. In the ChIP-qPCR assay (middle and right panels), chromatin immunoprecipitations were performed with either 10 µl of RING1B (D22F2) XP® Rabbit mAb #5694 or 5 µl of SUZ12 (D39F6) XP® Rabbit mAb #3737. The enriched DNA was quantified by real-time PCR using SimpleChIP® Mouse HoxD10 Exon 1 Primers #7429, SimpleChIP® Mouse HoxA1 Promoter Primers #7341, and SimpleChIP® Mouse GAPDH Intron 2 Primers #8986. The amount of immunoprecipitated DNA in each sample is represented as normalized signal to the negative GAPDH locus (equivalent to one).
Appendix C: Optimization of Chromatin Fragmentation
Optimal conditions for the fragmentation of cross-linked chromatin DNA is highly dependent on the number of cells, volume of sample, length of sonication, and sonicator power setting used. For each sonication sample, we recommend using 100-150 mg of tissue or 1 x 107 to 2 x 107 cells per 1 ml ChIP Sonication Nuclear Lysis Buffer. Below is a protocol for determination of the optimal sonication conditions for a specific tissue or cell type.
- Prepare cross-linked nuclei from 100 to 150 mg of tissue or 1 x 107 to 2 x 107 cells, as described in Sections I, II, and III. Stop after Step 4 of Section III and proceed as described below.
- Fragment chromatin by sonication. Optimal sonication conditions can be determined for a given sonicator by varying the number of rounds or duration of sonication at a given power setting (see Step 5 in Section III for optimal power setting using a Branson Digital Sonifier 250 probe sonicator). To determine optimal sonication conditions, set up a sonication time-course experiment and remove 50 µl samples of chromatin after a given round or duration of sonication. For example, take chromatin samples after each 1 to 2 min of sonication.
- Clarify chromatin samples by centrifugation at 21,000 x g in a microcentrifuge for 10 min at 4°C.
- Transfer supernatants to new microfuge tubes and add 100 µl nuclease-free water, 6 µl 5 M NaCl #7010, and 2 µl RNAse A #7013. Vortex to mix and incubate samples at 37°C for 30 min.
- To each RNase A-digested sample, add 2 µl Proteinase K #10012. Vortex to mix and incubate samples at 65°C for 2 hr.
- Remove 20 µl of each sample and determine DNA fragment size
- Choose the sonication conditions that generate optimal DNA fragment size and use for chromatin preparation in Step 5 of Section III. If optimal sonication conditions are not achieved, increase or decrease the power setting of the sonicator and repeat the sonication time course.
NOTE: Optimal sonication conditions can vary with different sample types and fixation times. Use the minimal number of sonication cycles required to generate the desired length of chromatin fragments. Over sonication, as indicated by more than 80% of total DNA fragments less than 500 bp, can result in excessive damage to the chromatin and result in lower immunoprecipitation efficiency (see Figure 8, right panel).
- For sonication of cells fixed for 10 min, optimal sonication conditions will generate a DNA smear with approximately 90% of total DNA fragments less than 1 kb (see Figure 8, left panel). Increasing the fixation time to 30 min will limit fragmentation, generating a DNA smear with approximately 60% of total DNA fragments less than 1 kb.
- For sonication of tissues fixed for 10 min, optimal sonication conditions will generate a DNA smear with approximately 60% of total DNA fragments less than 1 kb. Increasing the fixation time to 30 min will limit fragmentation, generating a DNA smear with approximately 30% of total DNA fragments less than 1 kb (see Figure 7, left panel).
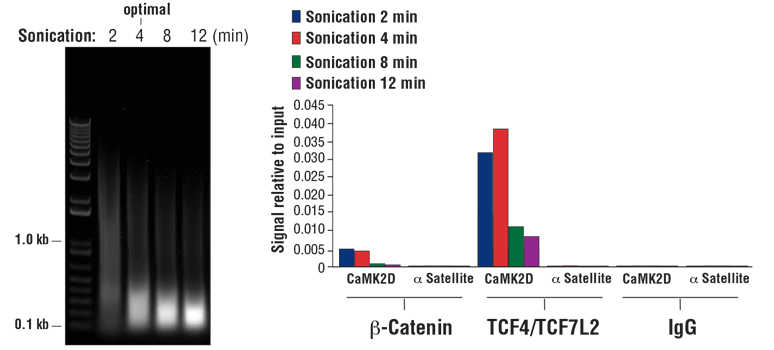
FIGURE 8. Chromatin immunoprecipitations were performed with 2 x 107 HCT 116 cells that were cross-linked for 10 min and sonicated for the time indicated (left panel). DNA was purified as described in Section IV of the SimpleChIP® Plus Sonication Chromatin IP Kit #56383, and 20 µl of purified DNA was separated by electrophoresis on a 1% agarose gel. As shown in the left panel, increasing cycles of sonication reduces the size of chromatin fragments. Chromatin immunoprecipitations were performed with either 5 µl of Non-phospho (Active) Β-Catenin (Ser33/37/Thr41) (D13A1) Rabbit mAb #8814, 10 µl of TCF4/TCF7L2 (C48H11) Rabbit mAb #2569, or 2 µl of Normal Rabbit IgG #2729 using SimpleChIP® Plus Sonication Chromatin IP Kit #56383. The enriched DNA was quantified by real-time PCR using SimpleChIP® Human CaMK2D Intron 3 Primers #5111 and SimpleChIP® Human α Satellite Repeat Primers #4486. The amount of immunoprecipitated DNA in each sample is represented as signal relative to the total amount of input chromatin (equivalent to one; right panel). As shown, 4 min of chromatin sonication is optimal when using a Branson Digital Sonifier D250 probe sonicator with a 1/8 inch Micro Tip. Over-sonication significantly impairs enrichment of both cofactor beta-catenin and transcription factor TCF4/TCF7L2 containing chromatin.
Appendix D: Troubleshooting Guide
| Problem |
Possible Causes |
Recommendation |
| 1. Concentration of the fragmented chromatin is too low. |
Cell/nuclear lysis is incomplete.
Not enough cells were used for the chromatin preparation.
|
If DNA concentration of the chromatin preparation is close to 50 µg/ml, add additional chromatin to each IP to give at least 5 µg/IP and continue with protocol.
Count a separate plate of cells before cross-linking to determine an accurate cell number.
|
Shorten the crosslinking time within 10-30 minute range. Reduce the amount of cell/tissues per sonication. Conduct sonication time course.
| 2. Chromatin is under-fragmented and fragments are too large (>50% above 1.5kb). |
Cells may have been over cross-linked.
Too many cells/tissues were processed.
|
Shorten the crosslinking time within 10-30 minute range. Reduce the amount of cell/tissues per sonication. Conduct sonication time course.
|
| 3. Chromatin is over-fragmented (>90% under 300 bp). |
Sonication condition is too harsh.
|
Conduct a sonication time course to find a minimum output/duration to achieve appropriate sonication.
|
| 4. No product or very little product in the input PCR reactions. |
Not enough DNA added to the PCR reaction or conditions are not optimal.
PCR amplified region may span nucleosome-free region.
Not enough chromatin added to the IP or chromatin is over-sonicated
|
Add more DNA to the PCR reaction or increase the number of amplification cycles.
Optimize the PCR conditions for experimental primer set using purified DNA from cross-linked and sonicated chromatin. For optimal ChIP results add 5-10 µg chromatin per IP. See recommendations for problems 1 and 3 above.
|
| 5. No product in the positive control Histone H3-IP RPL30 PCR reaction. |
Not enough chromatin or antibody added to the IP reaction or IP incubation time is too short.
Incomplete elution of chromatin from Protein G beads.
|
Be sure to add 5-10 µg of chromatin and 10 µl of antibody to each IP reaction and incubate with antibody over-night and an additional 2 hr after adding Protein G beads.
Elution of chromatin from Protein G beads is optimal at 65°C with frequent mixing to keep beads suspended in solution.
|
| 6. Quantity of product in the negative control Rabbit IgG-IP and positive control Histone H3-IP PCR reactions is equivalent. |
Too much or not enough chromatin added to the IP reaction. Alternatively, too much antibody added to the IP reaction.
Too much DNA added to the PCR reaction or too many cycles of amplification.
|
Add no more than 15 µg of chromatin and 10 µl of histone H3 antibody to each IP reaction. Reduce the amount of normal rabbit IgG to 1 µl per IP.
Add less DNA to the PCR reaction or decrease the number of PCR cycles. It is very important that the PCR products are analyzed within the linear amplification phase of PCR. Otherwise, the differences in quantities of starting DNA cannot be accurately measured.
|
| 7. No product in the Experimental Antibody-IP PCR reaction. |
Not enough DNA added to the PCR reaction.
Not enough antibody added to the IP reaction.
Antibody does not work for IP.
|
Add more DNA to the PCR reaction or increase the number of amplification cycles.
Typically a range of 1 to 5 µg of antibody is added to the IP reaction; however, the exact amount depends greatly on the individual antibody.
Increase the amount of antibody added to the IP. Find an alternate antibody source.
|
posted March 2017
revised April 2022

